Production Scale 
Description
This service allows you to best separate two sets of protein sequences, considering only the amino acid composition of these sequences. The method is recommended to be used if there is a relatively small set of sequences. Our method works if each set contains more than 20 sequences. Meanwhile, complex algorithms like neural networks have a hard time learning on sets of 1000 sequences. A detailed analysis of the algorithm is given in the article "Comparison of deep learning models with a simple method to assess the problem of antimicrobial peptides prediction" (forthcoming). We outline only the basic principles. The result of the program is a scale, each of the amino acids is assigned a certain value wk. Any protein sequence can be assigned
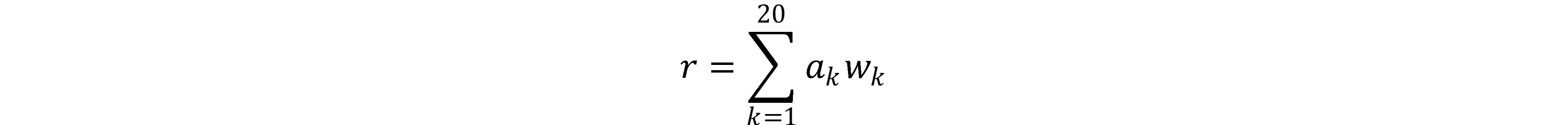
where ak is the fraction of each amino acid in the sequence. If r > 0, then we say that the given sequence is more similar to the sequences from the first set. If r < 0, then on the sequence from the second set. The quality of separation can be assessed by the following characteristics:
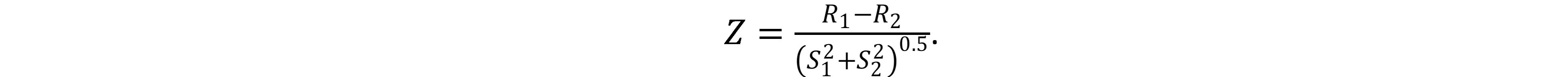
Z is the main optimized parameter. Here R1 and R2 are the mean values of r over sets 1 and 2. S1 and S2 are the standard deviation over these sets. It is interesting to note that at Z > 4 the bases are separated by 100%. Normal parameters stop improving, and Z can show better separation quality. Truth 1 is the proportion of correctly predicted from set 1, Truth 2 is from set 2. And the last estimate is AUC (area under the curve). AUC